The standard theory for how SARS-CoV-2 infects a cell is simple, too simple. The virus is surrounded by Spikes. One Spike connects with the ACE2 receptor, on the surface of the cell. Then TMPRSS2 cuts one of the proteins in the viral Spike, in two places, and the cell membrane merges with the viral membrane. Both membranes are lipid bilayers. The virus then drops its RNA and Nucleocapsid protein into the cytoplasm. And the virus uses the cell to make many copies of itself.
“One mechanism for SARS-CoV-2 entry occurs when the spike protein on the surface of SARS-CoV-2 binds to an ACE2 receptor followed by cleavage at two cut sites (“priming”) that causes a conformational change allowing for viral and host membrane fusion. This fusion event is proceeded by release of viral RNA within the host cell. TMPRSS2 has an extracellular protease domain capable of cleaving the spike protein to initiate membrane fusion. Additionally, knock-out studies in mice have demonstrated reduced infection in the absence of TMPRSS2” [1]
Notice that “knock-out studies in mice” show the infection is only reduced “in the absence of” the TMPRSS2 step — and not completely prevented. Infection can still occur without TMPRSS2. Even so, inhibiting that protease should reduce the infectivity and severity of Covid-19. So it is a viable approach, but one that needs to be accompanied by other inhibitors.
TMPRSS2 inhibitors are in trials for testing against Covid-19. The most promising are these two, which in vitro studies have verified as effective inhibitors: “nafamostat (IC50 = 0.27 nM), camostat (IC50 = 6.2 nM)” [1]. By a comparison of their IC50’s, nafamostat should be 23 times more effective than camostat. But that remains to be proven.
The process of infection supposedly begins with this docking of the SARS-CoV-2 Spike with ACE2. But the ACE2 protrudes out from the cell membrane, and the Spike protrudes out from the viral membrane. This docking is rather like an acrobat, doing a one-arm hand-stand on top of the other acrobat’s single raised arm. The high rate of infection does not match this seemingly very difficult process. The Spike has to dock with the ACE2, and there has to be a TMPRSS2 close by. And then the virus is much larger than any one Spike, and the Spikes are very flexible in their stalks:
“We show that the stalk domain of S contains three hinges that give the globular domain unexpected orientational freedom. We propose that the hinges allow S to scan the host cell surface, shielded from antibodies by an extensive glycan coat.” [2]
It seems that the Spike would not be capable of keeping the entire virus away from the cell wall and the ACE2 protein.
What this paper suggests, instead, is that the virus lands on the cell membrane, anywhere. Multiple virions land across the cell wall. Sometimes they move across the surface via “rafts” in the lipid bilayer. But more often, they move by rolling across the surface. The Spikes with their heavy glycan coating reduce friction with the cell surface. The Spikes themselves reduce the contact area, lessening resistance to movement, and the sugar chains have a paradoxical role, at once both helping the virus to remain attached to the cell surface, regardless of which surface proteins are between the virus and the cell membrane, and yet allowing enough flexibility for the virus to move.
In addition, the flexibility of the Spikes and their many sugar-chains extending in every direction allow the virions to stick to one another. This is seen clearly in many electron micrographs. In the first micrograph below, the virions seem to be able to tumble cross the cell surface and over other another. There is fluid around them, of course, but they don’t float away, as they can stick lightly to one another and to the surface. But not so much so that they can’t move.
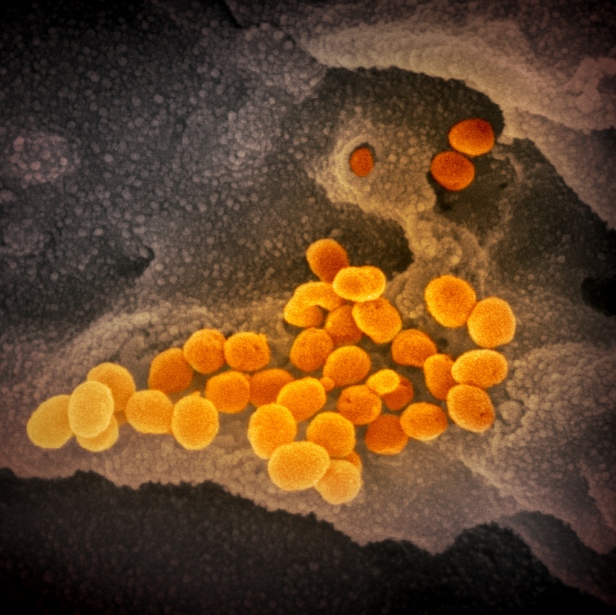
In the next micrograph below, the virions are clumped together. They plainly have the ability to stick to one another, and to seemingly any location on the cell surface.
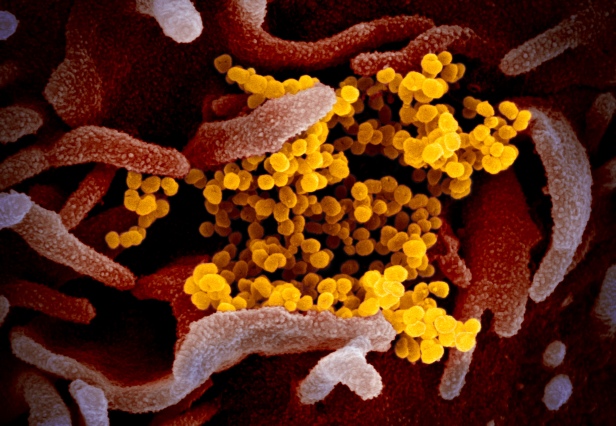
The third micrograph demonstrates both these points again. SARS-CoV-2 can attach to almost any location on a cell surface, using the flexibility of the Spikes and the ability of their glycans to act like Velcro® and stick to a wide range of cell surface proteins, as well as to one another.
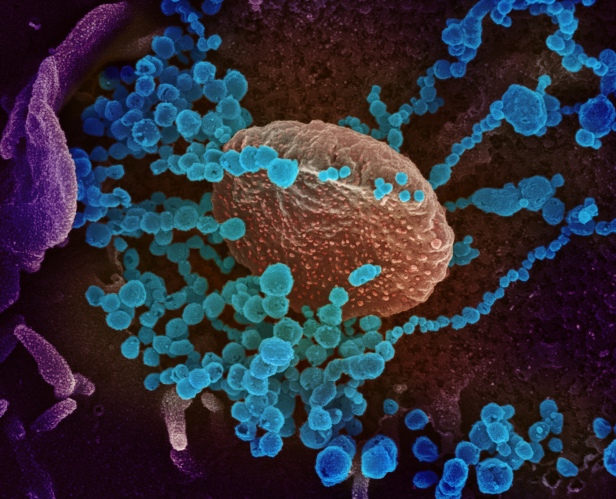
Infectivity
What this means for infectivity is that the virus need not “find” the ACE2 receptor right away. It can land on the surface of any cell, and stick there. Then it can roll across the surface and spend hours or days waiting for that connection with ACE2. Then the TMPRSS2 function is helpful, but does not seem to be essential. We know that it is possible for SARS-CoV-2 to infect cells which lack both ACE2 and TMPRSS2, through the CD147 protein [4]. Another article showed that cellular Heparan Sulfate, along with ACE2, can facilitate infection of cells by SARS-CoV-2 [5]. One study found that “SARS-CoV-2 surface proteins have better interactions with DPP4 and Meprin A alpha host cells receptors rather than ACE2 [6].” So the point here is that the infection of cells by SARS-CoV-2 does not occur in the simplistic manner that is usually described.
In the more complex explanation, offered by this paper, the virus is so highly infective because it can stick to the surface of almost any cell, and move across the surface, seeking a surface protein that it can use to infect the cell. ACE2 with TMPRSS2 is ideal for the virus, but other modes of entry are possible.
Now, the ability of the virus to stick to other viruses, and to clump together, may be a factor in its high infectivity. Once one virus begins membrane fusion, perhaps other viruses, stuck to the infecting virus, might be able to infect the cell in the same location. This would occur by endocytosis, when the cell membrane surrounds the virus that docks with the ACE2 protein, possibly also surrounding other virions that have attached themselves to the primary infecting virus particle.
Microthrombosis
SARS-CoV-2 can infect platelets, causing a hyper-activation of the platelets which increases clotting [7]. It can also infect the endothelial cells that line the blood vessels. The infection of endothelial cells soon destroys the cell, and said cellular destruction is a trigger for clotting [7].
Now, recall that SARS-CoV-2 can use CD147 to infect cells. Red Blood Cells (RBCs) have CD147 on the surface [8]. But in this case, the binding to the CD147 would not cause infection, as RBCs are rather dissimilar to other cells in the body. Rather, the Spike attaches to the CD147 protein, and remains on the outside of the RBC, then other spikes on the virus connect to other virions, and some of them attach to other RBCs in the same manner. Then the virus particles can attach to the outside of platelets and endothelial cells, causing a blood clot. This would be most likely to occur in the smallest blood vessels, the capillaries, as there is so little space in the vessel. The virus can easily cause a clot in a confined location.
Capillaries are 50 to 10 µmeters in diameter, which is 5,000 to 10,000 nanometers. SARS-CoV-2 virions are 50 to 200 nanometers in diameter. So the smallest capillaries are 25 to 100 viruses in diameter, and the largest are 50 to 200 viruses in diameter. When the virus attaches to RBCs, which are 6,000 to 8,000 nanometers in diameter (and often have to bend to fit through capillaries), not many viruses are needed to clog a capillary. If a set of virions, clump together, and then join two or three RBCs together, they can easily begin to clog capillaries. And then the clot attaches to epithelial cells via the connection of the Spikes to the cell walls and the RBCs. Infection of epithelial cells releases clotting factors from the cells, and now fibrin solidifies the clot.
This process can cause a cascade of virus particles clotting with one another and with RBCs, and then setting off the destruction of epithelial cells that releases more fibrin. Soon the clotting becomes massive, across innumerable capillaries in an organ, shutting it down. Multi-organ failure is one of the common causes of death via Covid-19.
This theory of the SARS-CoV-2 virus connecting to RBCs and epithelial cells is found in:
Scheim, David. “Antimalarials for COVID-19 treatment: rapid reversal of oxygen status decline with the Nobel Prize-honored macrocyclic lactone ivermectin.” Available at SSRN 3617911 (2020). Link to Study
He describes the theory in this way:
“The central role of the CD147 transmembrane receptor in the binding of SARS-CoV-2 is considered. A catch and clump scenario for impedance of capillary flow through viral bindings to blood cells via CD147 is proposed as a possible explanation for the observed rapid clinical response to IVM and for other puzzling aspects of COVID-19.”
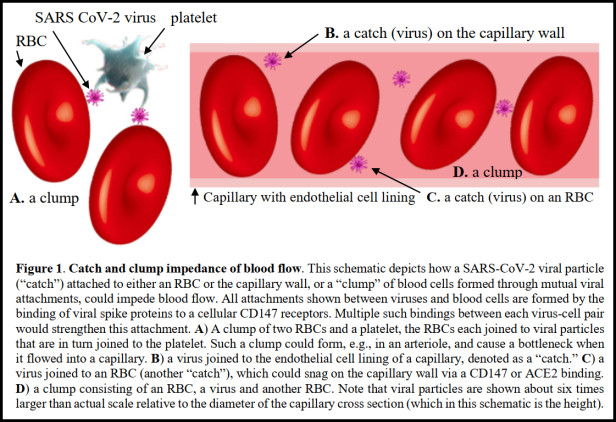
Summary
This article suggests a new theory about the process of the infection of cells by SARS-CoV-2. The process involves a proposed ability of the virus to stick to the cell membrane, using the glycan-coated Spikes, and to move across the cell surface to find ACE2 and TMPRSS2 surface proteins. The ability of the virus to stick to other viruses and clump together is also a factor in this process. If the virus can stick to other viruses in this manner, then the theory of David Scheim that the viruses clump together with RBCs to form clots makes sense. This explains the problem of microthrombosis found in many hospitalized Covid-19 patients.
What can be done to prevent microthrombosis and also reduce infectivity, if the above theories are correct? ACE2 receptors can be protected with natural inhibitors, that bind to the ACE2 and prevent the Spikes from docking with the receptor. Then other natural inhibitors exist which bind to the Spike protein, also preventing docking with ACE2.
It is an open question as to whether inhibitors on the Spikes will affect thrombosis. Some in vitro studies would need to be done to determine the effects of these ligands on the ability of SARS-CoV-2 to form clots.
Ronald L. Conte Jr.
Covid.us.org
an author, not a doctor
ENDNOTES:
1. Shrimp, Jonathan H., et al. “An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19.” ACS Pharmacology & Translational Science (2020).
2. Turonová, Beata, et al. “In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges.” Science (2020).
3. Sundari, C. Sivakama, B. Raman, and D. Balasubramanian. “Hydrophobic surfaces in oligosaccharides: Linear dextrims are amphiphilic chains.” Biochimica et Biophysica Acta (BBA)-Biomembranes 1065.1 (1991): 35-41.
4. Wang, Ke, et al. “SARS-CoV-2 invades host cells via a novel route: CD147-spike protein.” BioRxiv (2020).
5. Clausen, Thomas Mandel, et al. “SARS-CoV-2 Infection depends on cellular Heparan Sulfate and ACE2.” Cell (2020).
6. Goothy, Sai Sailesh Kumar, and Arun HS Kumar. “Network Proteins of Angiotensin-converting Enzyme 2 But Not Angiotensin-converting Enzyme 2 itself are Host Cell Receptors for SARS-Coronavirus-2 Attachment.” BEMS Reports 6.1 (2020).
7. Zaid, Younes, et al. “Platelets can contain SARS-CoV-2 RNA and are hyperactivated in COVID-19.” medRxiv (2020).
8. Scheim, David. “Antimalarials for COVID-19 treatment: rapid reversal of oxygen status decline with the Nobel Prize-honored macrocyclic lactone ivermectin.” Available at SSRN 3617911 (2020).
